科技日报记者 刘园园
记者4月2日从西湖大学获悉,该校杨剑教授研究团队与合作者开发出基于泛基因组的联合组装(PIGA)新方法,成功构建了目前全球规模最大的人类泛基因组,并发现人类基因组中约13%的未知序列,为遗传疾病诊断与精准医疗提供了关键支撑。
人类基因组被誉为生命地图,自2003年人类基因组计划完成后,科学研究长期使用单一参考基因组作为研究标准。但是,这一“标准地图”存在大量盲区,难以适配个体间遗传差异。而传统泛基因组研究受限于高昂的三代测序成本,样本量多在几十至百人,难以全面反映人类遗传多样性。
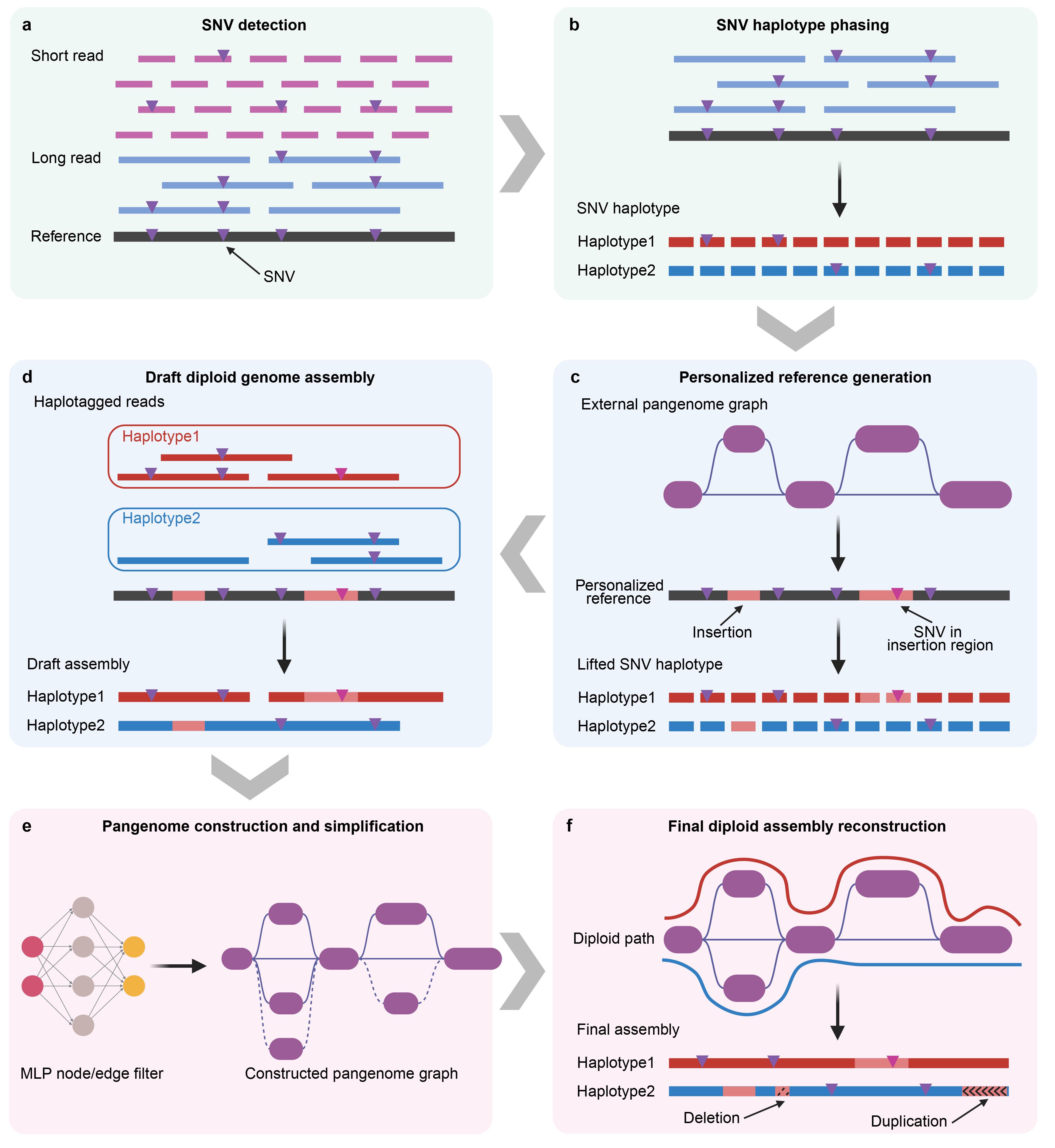
为破解这一困境,杨剑团队提出采用高性价比的二代与三代混合测序策略,创新性地开发出一套基于泛基因组的联合组装(PIGA)方法。其中,二代测序技术保障序列准确性,三代测序技术搭建基因组骨架,该方法在提升测序质量的同时,还显著降低了测序成本。依托该方法,团队成功构建包含1116个二倍体基因组的泛基因组,错误率仅五万分之一,实现了大规模人群高质量基因组组装。
“大样本量泛基因组可以极大地赋能罕见病诊断,尤其是针对那些之前未知的复杂变异。”杨剑解释道,只有样本量足够大,才能准确估计某个复杂变异在人群中的频率。
研究显示,传统参考的人类基因组遗漏了超过4亿碱基对的未知序列,相当于人类基因组总规模的13%。研究团队在新发现序列中识别出2620万碱基对的功能基因与调控元件,这些元件可指导蛋白质合成并调控基因表达。同时,他们绘制出迄今最详尽的人类遗传变异图谱,涵盖3540万个小变异,还破译了大量难以检测的结构变异、串联重复序列、嵌套变异等复杂变异。此外,研究团队成功定位了3256个调控基因表达的关键复杂变异,为疾病遗传诊断提供重要依据。
“这项研究极大地深化了我们对复杂遗传变异及其功能的认知,彰显了基于大规模基因组组装的分析方法在填补传统测序研究空白方面的巨大潜力,为人类健康研究和其他物种泛基因组研究提供了新范式。”杨剑说。
 网友评论
网友评论